FermiNet(フェルミネット):第一原理に基づく量子物理学と化学
FermiNet Quantum physics and chemistry based on first principles.
最近、『Physical Review Research』に掲載された記事では、ディープラーニングが現実世界のシステムの量子力学の基本方程式を解くのに役立つことを示しています。これは重要な基礎科学の問いでもあり、将来的には実用的な利用方法につながる可能性もあります。これにより、研究者は実験室で作成する前に、シリコン上で新しい材料や化学合成をプロトタイプ化することができます。本日、この研究からのコードも公開しましたので、計算物理学や化学のコミュニティは私たちの研究を元にさまざまな問題に取り組み、応用することができます。私たちは、量子力学の基本的な問題を解決するための、DeepMindのAI研究で開発されたツールやアイデアが自然科学の分野で重要な問題を解決するのに役立つことを願っています。また、FermiNetは、タンパク質の折りたたみ、ガラスのダイナミクス、格子量子クロモダイナミクスなどのプロジェクトとともに、そのビジョンを具現化するための私たちの取り組みに加わりました。
量子力学の簡単な歴史
「量子力学」という言葉を出すと、混乱を引き起こす可能性があります。このフレーズは、シュレディンガーの猫のイメージを思い浮かべさせます。猫は生きていると死んでいるの両方の状態を同時に持つことができる、そして、何らかの方法で波でもある基本的な粒子です。量子系では、電子などの粒子は、古典的な記述では正確な位置を持たず、その位置は確率的なクラウドで表されます。つまり、それは許されるすべての場所に広がっています。この直感に反する状態が、リチャード・ファインマンをして「量子力学を理解していると思っているなら、それは量子力学を理解していない」と宣言させるほどでした。この奇妙な状態にもかかわらず、この理論の中身はわずかな数式にまとめることができます。その中で最も有名なのが、シュレディンガー方程式で、量子スケールの粒子の振る舞いを、ニュートンの法則が私たちのより馴染みのある人間のスケールの物体の振る舞いを記述するように記述します。この方程式の解釈は、頭を悩ませることがありますが、数学的にははるかに扱いやすく、教授からは「黙って計算しろ」というよくある忠告が生まれました。
これらの方程式は、私たちが周りで見るすべての物質の振る舞いを原子や核のレベルで説明するのに十分です。その直感に反する性質が、超伝導体、超流体、レーザー、半導体などのエキゾチックな現象をもたらします。しかし、単なる共有結合という一見地味な結合も、電子の量子相互作用の結果です。これらのルールが1920年代に解明されると、科学者たちは初めて、化学がどのように機能するかについての詳細な理論を持つことになりました。原理的には、異なる分子のためにこれらの方程式を設定し、系のエネルギーを解き、どの分子が安定していて、どの反応が自発的に起こるかを把握することができます。しかし、これらの方程式の解を実際に計算しようとすると、最も単純な原子(水素)の場合には正確に求めることができるものの、それ以外のほとんどの分子では計算が複雑すぎました。
当時の楽観主義は、ポール・ディラックによってうまくまとめられました。
物理学の大部分および化学全体の数学的理論に必要な基礎的な物理法則は完全に知られている。問題は、これらの法則の正確な適用が、解けないほど複雑な方程式につながることである。したがって、量子力学の近似的な実用的な方法の開発が望まれる。 ポール・ディラック, 1929年
これに呼応して、多くの人々が行動を開始し、物理学者たちは分子の結合や他の化学現象の定性的な振る舞いを近似的に記述する数学的な手法を構築しました。これらの手法は、入門的な化学でおなじみの電子の振る舞いのおおよその説明から始まります。この説明では、各電子は特定の軌道に割り当てられ、原子核の近くの任意の点で単一の電子が見つかる確率を与えます。各軌道の形状は、すべての他の軌道の平均的な形状に依存します。この「平均場」の説明では、各電子は1つの軌道に割り当てられているとして扱われるため、電子の実際の振る舞いの非常に不完全なイメージです。それにもかかわらず、これは分子の総エネルギーを約0.5%の誤差で推定するのに十分です。
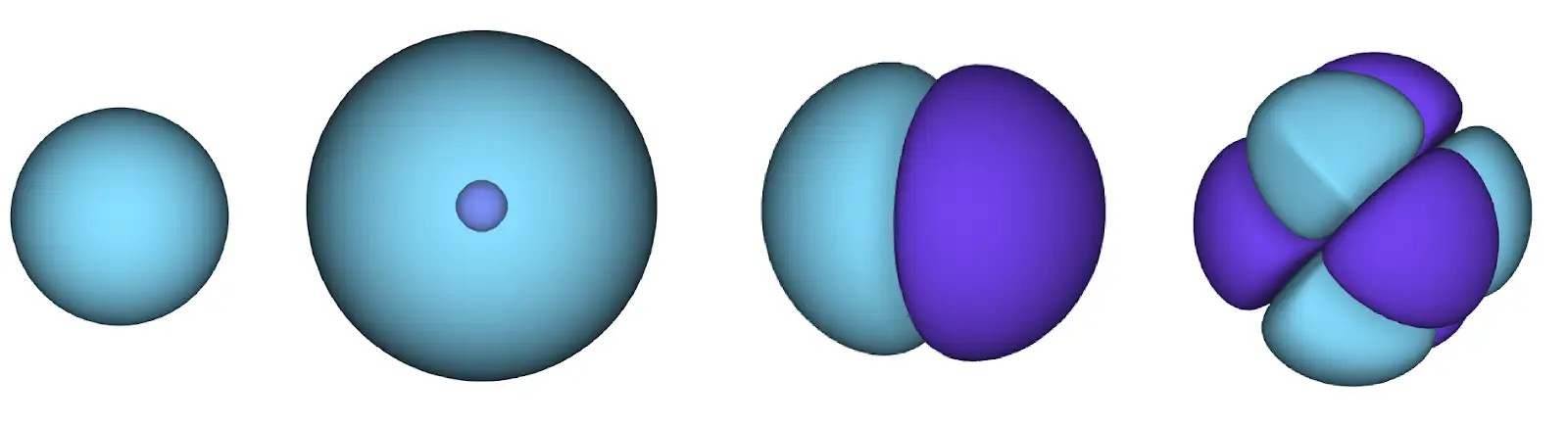
残念ながら、0.5%の誤差では、実際の化学者にとって有用ではありません。分子結合のエネルギーは、システム全体のエネルギーのごく一部であり、分子が安定しているかどうかを正確に予測するためには、システム全体のエネルギーの約0.001%、または残りの「相関」エネルギーの約0.2%に依存することがよくあります。たとえば、ブタジエン分子の電子の総エネルギーはほぼ100,000キロカロリー/モルですが、分子の異なる可能な形状の間のエネルギー差はわずか1キロカロリー/モルです。つまり、ブタジエンの自然な形状を正確に予測するには、フットボール場の幅をミリメートル単位で測定するのと同じレベルの精度が必要です。
第二次世界大戦後のデジタルコンピューティングの登場により、科学者たちは電子の平均場記述を超えるさまざまな計算方法を開発しました。これらの方法は、略語のアルファベットスープで提供されますが、一般的には精度と効率のトレードオフを行う軸のどこかに位置しています。一方では、ほぼ正確なメソッドがありますが、電子の数に対して指数関数的に拡大し、小さな分子以外では実用的ではありません。他方では、線形に拡大するが、あまり正確ではありません。これらの計算方法は、化学の実践に非常に大きな影響を与えました – 1998年のノーベル化学賞は、これらのアルゴリズムの創始者に授与されました。
フェルミオニックニューラルネットワーク
既存の計算量子力学ツールの幅広さにもかかわらず、効率的な表現の問題に対処するために新しいメソッドが必要だと考えました。最も近似的な方法でも、最大の量子化学計算は数万個の電子にしか対応できない理由がありますが、分子動力学などの古典的な化学計算技術は数百万個の原子を扱うことができます。古典系の状態は簡単に記述できます – 各粒子の位置と運動量を追跡するだけです。量子系の状態を記述するのははるかに困難です。電子の位置のすべての可能な配置に確率を割り当てる必要があります。これは波動関数にエンコードされ、すべての電子の配置に対して正または負の数を割り当て、波動関数の2乗はその配置でシステムを見つける確率を与えます。すべての可能な配置の空間は膨大です – 各次元に100のポイントを持つグリッドとしてそれを表現しようとすると、ケイ素原子の可能な電子配置の数は宇宙の原子の数よりも大きくなります!
これが私たちが深層ニューラルネットワークが役立つと考えた理由です。ここ数年で、ニューラルネットワークを使用して複雑な高次元の確率分布を表現する方法に大きな進歩がありました。これらのネットワークを効率的かつスケーラブルにトレーニングする方法をすでに知っています。私たちは、これらのネットワークが人工知能の問題で高次元の関数に適合する能力をすでに証明しているので、量子波動関数を表現するためにも使用できるかもしれないと考えました。私たちは最初にこの考えを思いついたわけではありませんでした – Giuseppe CarleoやMatthias Troyerなどの研究者は、現代のディープラーニングを用いて理想化された量子問題を解決するために使用できることを示しています。私たちは、ディープニューラルネットワークを使用して、化学や凝縮系物理学のより現実的な問題に取り組むことを望んでおり、それには電子を計算に含める必要がありました。
ただし、電子を扱う場合には1つの注意点があります。電子はパウリの排他原理に従う必要があり、同じ空間に同時に存在することはできません。これは電子を含む粒子の一種であるフェルミオンに起因します。彼らの波動関数は反対称でなければなりません – 2つの電子の位置を入れ替えると、波動関数は-1倍されます。つまり、2つの電子が重なっている場合、波動関数(およびその構成の確率)はゼロになります。
これは私たちが、入力に対して反対称な新しいタイプのニューラルネットワークを開発する必要があったことを意味します。私たちはそれをフェルミオニックニューラルネットワークまたはFermiNetと名付けました。ほとんどの量子化学メソッドでは、反対称性は行列式と呼ばれる関数を使用して導入されます。行列式の特性は、2つの行を交換すると、出力が-1倍されることです。つまり、1つの電子関数の群を取り、システム内のすべての電子に対してそれらを評価し、すべての結果を1つの行列にまとめることができます。その行列の行列式は、適切に反対称な波動関数です。このアプローチの主な制限は、得られる関数(スレーター行列式として知られる)があまり一般的ではないことです。実際のシステムの波動関数は通常はるかに複雑です。これを改善する典型的な方法は、スレーター行列式の大規模な線型結合を取り、電子のペアに基づいたいくつかの単純な修正を追加することです。それでも、これでエネルギーを正確に計算するには十分ではない場合があります。
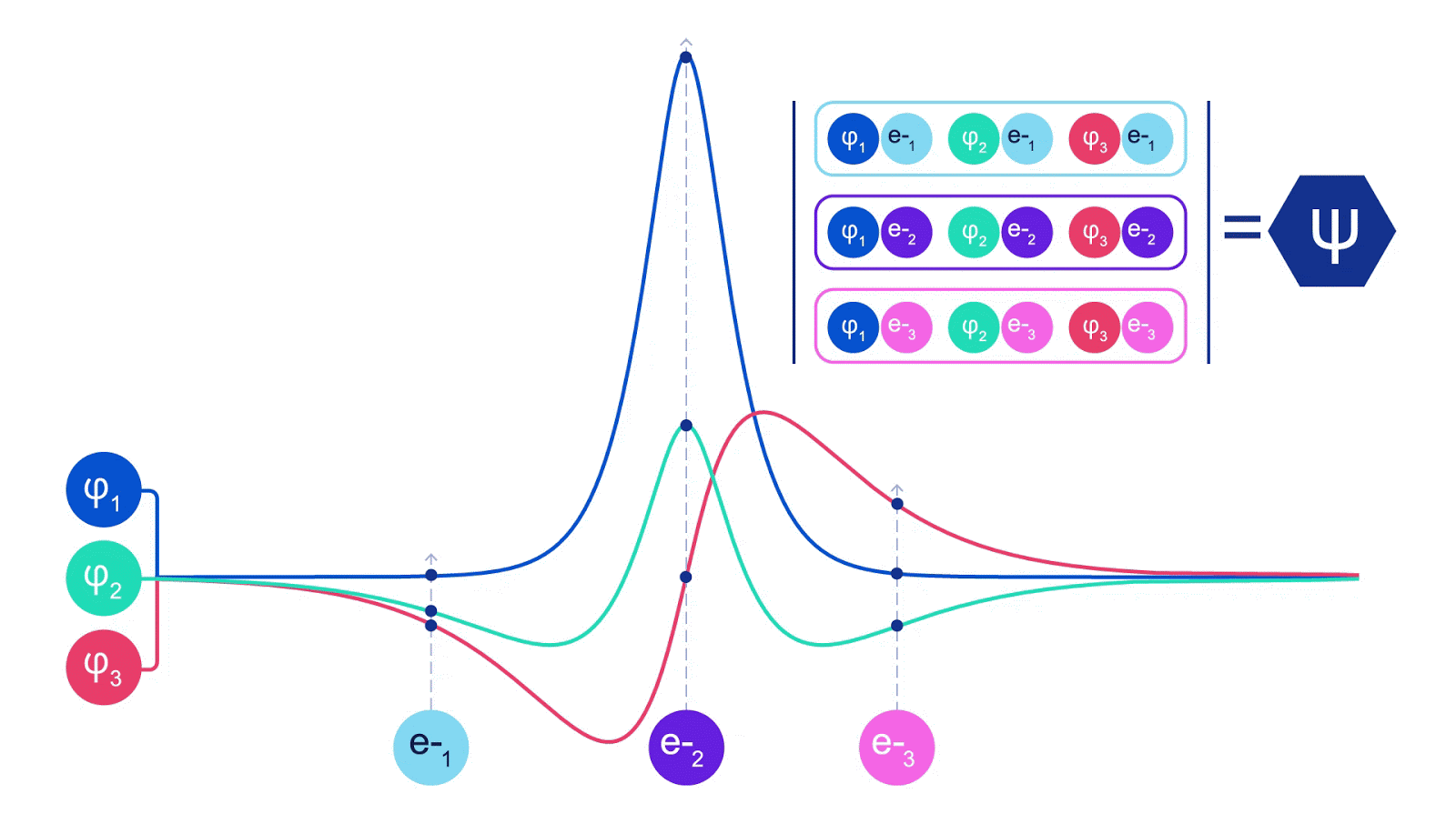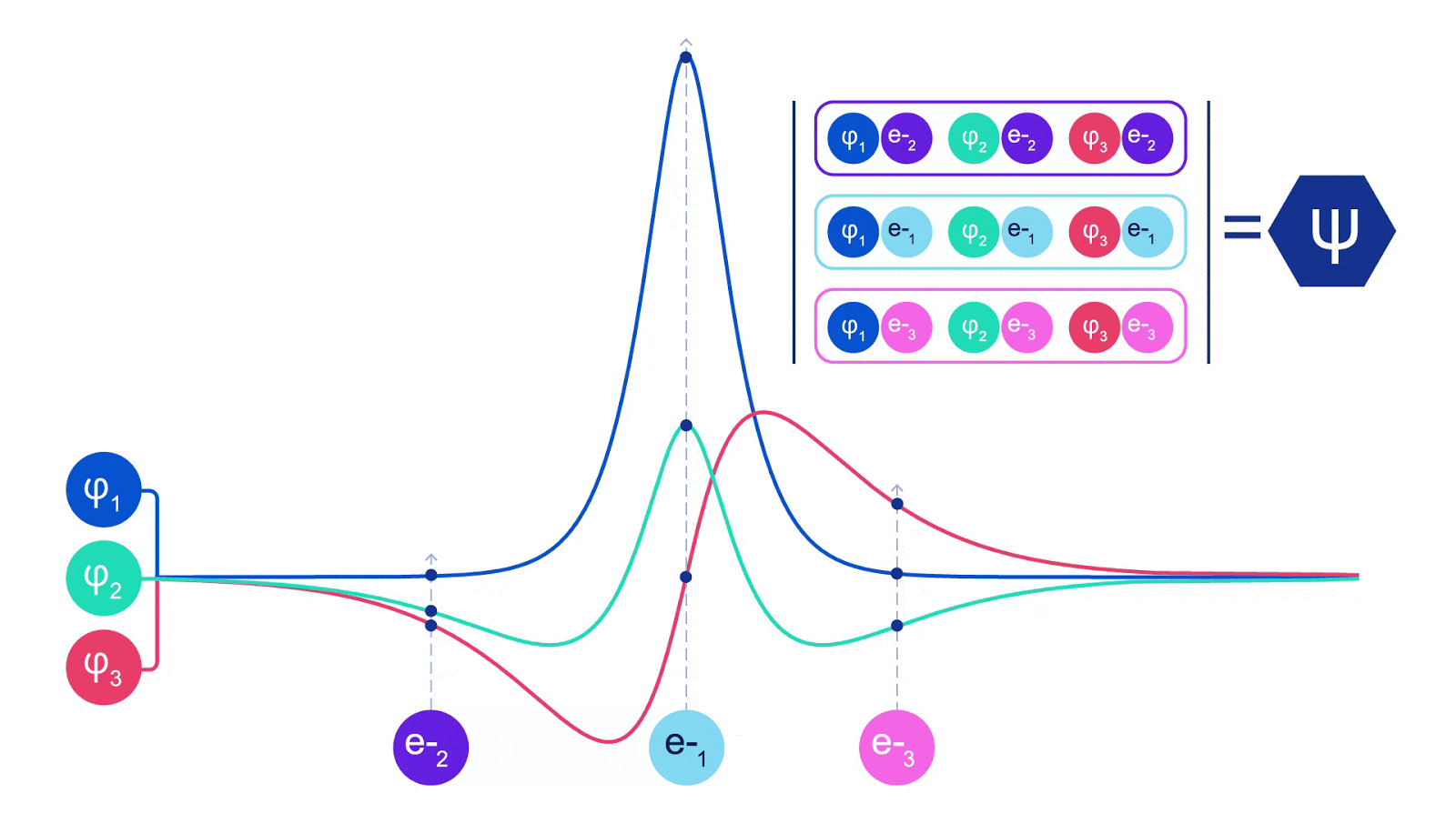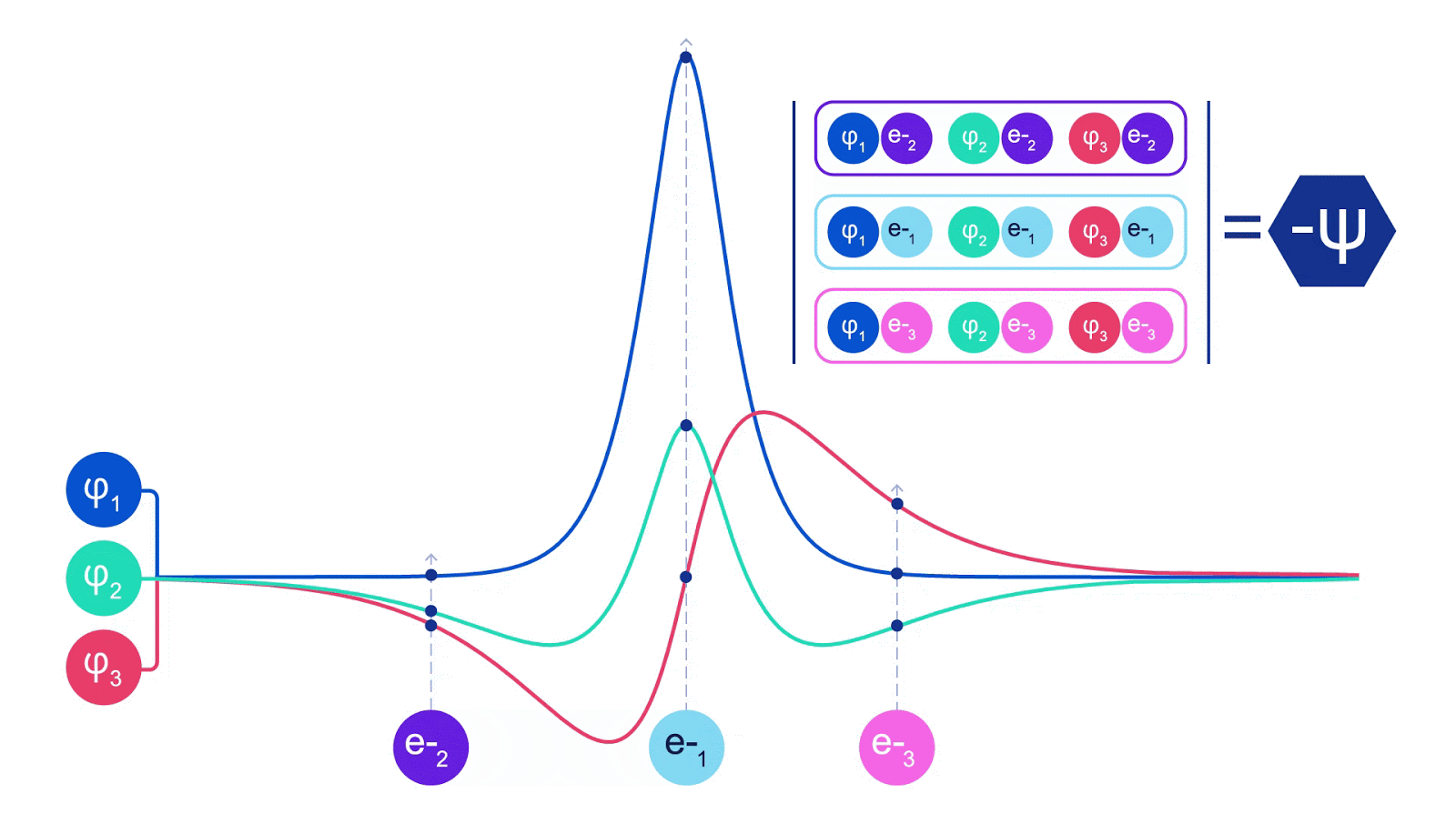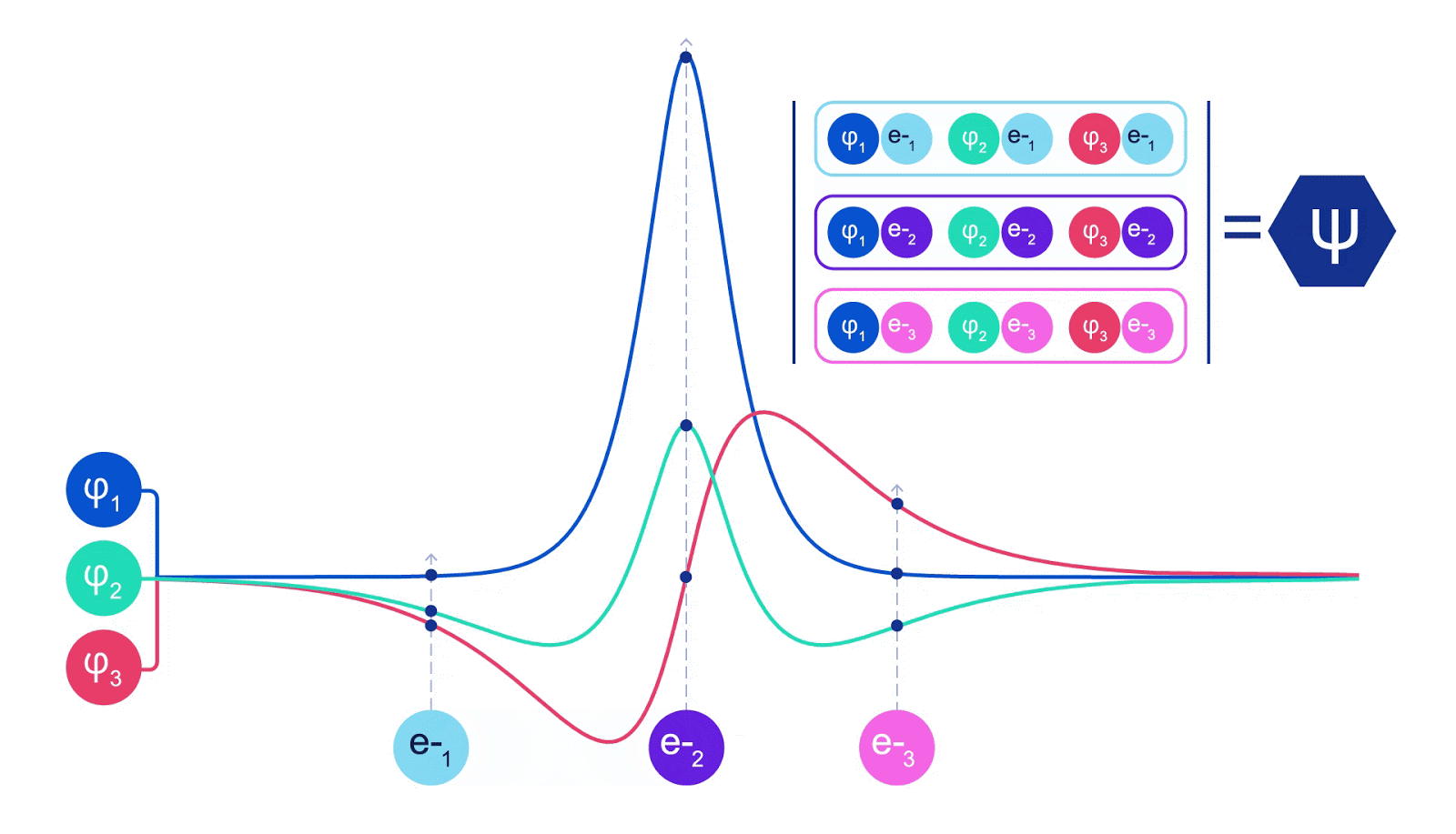
ディープニューラルネットワークは、しばしば基底関数の線形結合よりも複雑な関数を効率的に表現することができます。フェルミネットでは、スレーター行列式に入る各関数をすべての電子の関数にすることでこれを実現しています(1)。これは、単一および二電子関数のみを使用する方法をはるかに超えています。フェルミネットには、各電子ごとに個別の情報ストリームがあります。これらのストリーム間に相互作用がない場合、ネットワークは従来のスレーター行列式と同様の表現力しか持ちません。これを超えるために、ネットワークの各層で情報をストリーム全体にわたって平均化し、次の層の各ストリームにこの情報を伝えます。これにより、これらのストリームは反対称関数を作成するための正しい対称性を持つようになります。これはグラフニューラルネットワークが各層で情報を集約する方法と似ています。スレーター行列式とは異なり、フェルミネットは少なくともニューラルネットワークの層が十分に広くなる限り、普遍的な関数近似器です。つまり、これらのネットワークを正しくトレーニングできれば、シュレディンガー方程式のほぼ正確な解にフィットさせることができるはずです。
フェルミネットはシステムのエネルギーを最小化することでフィットさせます。それを正確に行うためには、電子のすべての可能な配置で波動関数を評価する必要があるため、代わりに近似的に行う必要があります。ランダムな選択の電子配置を選び、各電子配置ごとに局所的にエネルギーを評価し、各配置からの寄与を合計し、真のエネルギーの代わりにこれを最小化します。これはモンテカルロ法として知られています。それはまるでギャンブラーが何度もサイコロを振るようなものです。これは近似的な手法ですが、より正確にする必要があれば、サイコロを再び振ることができます。波動関数の2乗は、任意の位置で粒子の配置を観測する確率を与えるため、波動関数自体からサンプルを生成することが最も便利です。つまり、粒子の観測をシミュレートすることです。ほとんどのニューラルネットワークは、外部データからトレーニングされますが、私たちの場合、ニューラルネットワークでトレーニングに使用する入力は、ニューラルネットワーク自体によって生成されます。それは自分自身を引っ張って上に上がるようなものであり、電子が踊る原子核の位置以外のトレーニングデータは必要ありません。基本的なアイデアである変分量子モンテカルロ(またはVMCとしても知られる)は、60年代から存在しており、システムのエネルギーを計算するための安価ですがあまり正確ではない方法とされています。スレーター行列式に基づく単純な波動関数をフェルミネットに置き換えることで、私たちは見てきたすべてのシステムでこのアプローチの正確さを劇的に向上させました。
フェルミネットが現在の最先端の進歩を本当に表しているかどうかを確認するために、私たちは最初の周期表の原子(水素からネオンまで)などの単純でよく研究されたシステムを調査しました。これらは10個以下の電子を持つ小さなシステムであり、最も正確な(ただし指数関数的にスケーリングする)方法で扱うことができるほど単純です。フェルミネットは、比較可能なVMC計算と比較して、エラーを半分以上削減することが多く、指数関数的にスケーリングする計算に対するエラーを大幅に削減します。より大きなシステムでは、指数関数的にスケーリングする方法は扱いにくくなるため、代わりに「カプルドクラスター」法をベースラインとして使用します。この方法は、安定した構造を持つ分子ではうまく機能しますが、化学反応を理解する上で重要な結合の伸びや切れ目に苦しむことがあります。指数関数よりもはるかにスケーリングが良いですが、使用した特定のカプルドクラスター法は、電子の数を7乗した数にスケーリングするため、VoAGIサイズの分子にしか使用できません。私たちは、リチウム水素化物から始まり、30個の電子を持つ最大のシステムであるビシクロブタンまで、徐々に大きな分子にフェルミネットを適用しました。最小の分子では、フェルミネットはカプルドクラスターエネルギーと単一のスレーター行列式から得られるエネルギーとの差の99.8%を驚異的に捉えました。ビシクロブタンでは、フェルミネットはこの相関エネルギーの97%以上を捉えました。これは、「安価で正確ではない」アプローチにとっては非常に大きな成果です。
カプルドクラスターメソッドは安定した分子に対してはうまく機能しますが、計算化学の真のフロンティアは分子の伸縮、ねじれ、切れ目を理解することです。そこでは、カプルドクラスターメソッドはしばしば苦労するため、一貫した答えを得るために可能な限り多くの基準と比較しなければなりません。私たちは2つのベンチマークとなる伸縮したシステム、窒素分子(N2)と10個の原子からなる水素鎖(H10)を調査しました。窒素は特に難しい分子結合です。なぜなら、各窒素原子は3つの電子を寄与するからです。一方、水素鎖は、材料中で電子の振る舞いを理解するために興味があります。例えば、材料が電気を伝導するかどうかを予測することです。両方のシステムで、カプルドクラスター法は平衡状態ではうまくいきましたが、結合が伸びるにつれて問題が生じました。従来のVMC計算は全体的にはうまくいきませんでした。しかし、フェルミネットは、結合の長さに関係なく、調査された方法の中で最も優れたものの一つでした。
結論
ディープラーニングと計算量子化学の融合において、私たちはフェルミネットが素晴らしい進展の始まりだと考えています。私たちがこれまでに調査したほとんどのシステムはよく研究され、理解されています。しかし、他の分野におけるディープラーニングの最初の良い結果が追加の研究と急速な進歩をもたらしたように、私たちはフェルミネットがスケーリングアップとさらに優れたネットワークアーキテクチャのための多くの研究とアイデアをインスパイアすることを望んでいます。すでに、私たちの作業をarXivに初めて公開してから、他のグループも多電子問題に対する第一原理計算にディープラーニングを適用するアプローチを共有しています。また、私たちは計算量子物理学の表面しか触れていませんし、材料科学や凝縮物理学の難解な問題にフェルミネットを適用することを楽しみにしています。私たちの実験で使用したソースコードを公開することで、私たちの作業をベースに研究を進め、私たちがまだ夢見ていない新しいアプリケーションを試してみるような他の研究者をインスパイアできればと思っています。
We will continue to update VoAGI; if you have any questions or suggestions, please contact us!
Was this article helpful?
93 out of 132 found this helpful
Related articles




